Milos Cigoj firmly believes that everyone involved in the design of medical devices should keep in mind from the beginning (ideation and discovery) that having a solid submission strategy and a strong trail of records proving no step was skipped in the product evolution process is an imperative.
As a compliance and quality lead at HTEC, Milos has an outstanding experience in helping evangelise the importance of quality and compliance in medical device software development. He joined HTEC to, as he explains, “help HealthTech ecosystems overcome hurdles ahead before starting to smooth the path for disruptive medical technologies and benefit patients globally.”
We caught up with him to talk about why manufacturers frequently fail to bring novel devices to market and why HTEC plays a major role here as an innovation enabler.
Why is regulatory compliance an imperative on the medical innovation journey?
“Let me begin by saying that there is no single regulatory requirement; rather, everything is based on two concepts: The first is privacy, which includes the use of data for diagnosis, prevention, treatment, research, electronic data sharing, and other purposes. The second and most important factor is the risk-benefit ratio, which states that the medical benefits of using the device outweigh any potential risk – if we make mistakes, people could get hurt”, explains Milos.
Milos emphasises that everyone involved in the design of medical devices should keep in mind from the beginning (ideation and discovery) that there is no way around having a solid submission strategy and a strong trail of records proving no step was skipped in the evolution from idea to prototypes to final device.
“Medical device manufacturers must truly embrace the concept that the device dossier is the product, and the actual device is just a side effect”, says Milos.
Why do manufacturers frequently fail to bring a novel device to market?
It is common knowledge in the industry that the most frequent reasons for failure are:
- incorrect initial intended use and indications for use followed by a lack of proper engagement by potential users
- a lack of a clear market need
- poor value proposition articulation
- limited integration possibilities into existing clinical workflows
Milos points out that “If the exercise of addressing the previously mentioned pitfalls is not done upfront, other potential problems, such as a lack of a clear distribution and reimbursement strategy, and an inadequate team structure, could be right around the corner. All of these factors contribute to a lack of design control procedures, shaky facts and evidence-gathering activities, technical debt, rework, and a negative user experience. It is undoubtedly difficult if you are a startup embarking on this adventure without prior experience. There is little chance of a favorable outcome — the company going out of business as a result of burning all the money before even engaging with regulatory bodies is a looming possibility. Working with HTEC is unquestionably less expensive than going it alone. Compliance with stringent regulations is difficult for newcomers.
What sets the FDA apart on MedTech innovation?
According to a BCG survey, 79% of respondents agree that the FDA has responded effectively to advances in MedTech innovations over the past ten years. When compared to its international peers, FDA stands out for a number of reasons.
“I have a few key points to make here. When the FDA in the United States is compared to the EMA, MHRA, or other agencies in Europe and Asia, it is clear that the FDA, with the assistance of other agencies from the United States Department of Health and Human Services, is actually enabling and sponsoring innovation while others stand still. In other words, manufacturers now have more reliable, cost-effective market access in the United States. Manufacturers in the United States must demonstrate that their device is substantially equivalent to another legally sold device in the United States, known as a predicate. As the predicate, manufacturers must demonstrate that the device is safe to use and effective. As a result, the bar is raised for performing clinical trials, which are time-consuming, expensive, and involve multiple stakeholders”, says Milos.
On the other hand, Milos explains that, in Europe, producers must show exact equivalence, which begins with the exact intended use, indications for use, materials, production procedures, and even algorithms if we are talking about software. He points out that there is no other way to accomplish this without access to a technical file that the competition is unwilling to share. In short, this protects incumbent market participants while impeding new ones from becoming innovative. Other routes constitute a time-consuming and costly application process for the company.
“To summarize, the US Food and Drug Administration encourages innovation, whereas the European Union inhibits it. Due to tight rules, Europe appears to lag considerably behind medical innovation in the United States”, says Milos.
Are european medical device regulations limiting or fostering innovation?
Apart from HIPPA and FDA, new European medical device regulations, such as MDR and IVDR, seem to put even more pressure on startups as they have difficulty grasping regulatory compliance.
“Demand and supply in Europe are completely unbalanced. It’s considerably worse with MDR. In Europe, there are approximately forty recognized entities that can certify medical devices and less than ten that can assist in-vitro medical device submissions. It is considerably worse in the Software as a Medical Device scenario, where just a few Notified Bodies are accredited to analyze software. So, it’s a disaster, and it affects prices and timetables.”
According to the BCG survey, roughly half of the devices that could be put on the market with the now obsolete regulatory framework had been introduced in EU markets. Furthermore, 89% of corporations who sponsor these products say they will prioritize US regulatory approval in the future. Respondents also see new MDR/IVDR rules as complex and unpredictable, making it less compelling to develop and commercialize novel medical devices in Europe.
Building better regulatory compliance with HTEC
HTEC is the primary innovation facilitator here due to our extensive experience working on healthcare and life science projects over the years. HTEC can help startups and HealthTech ecosystems overcome hurdles ahead before starting up to smooth the path for new medical technologies and benefit patients globally.
“Our professionals have extensive domain knowledge and can assist clients from the product concept phase to the post-market phase. We assist the client in steering the design and manufacturing protocols and submission strategy by double-checking the technical and use-case feasibility in relation to the envisioned intended use and indications for use. Once the client is empowered to make an informed decision, we can augment the client’s design team in designing and validating prototypes, implementing the quality management system, including the information governance management aspect, developing the device dossier and the actual candidate device, performing pre-clinical studies, and engaging with authorities. As with medical devices, the product life cycle is truly a cycle. We also assist in post-market activities and improve designs for new device evolutions based on actual usage.
Based on my experience, I am confident that we can assist companies in avoiding high costs by focusing their efforts on developing something with tangible medicinal benefits, viable real-life use cases, and healthcare workflow integration possibilities.”
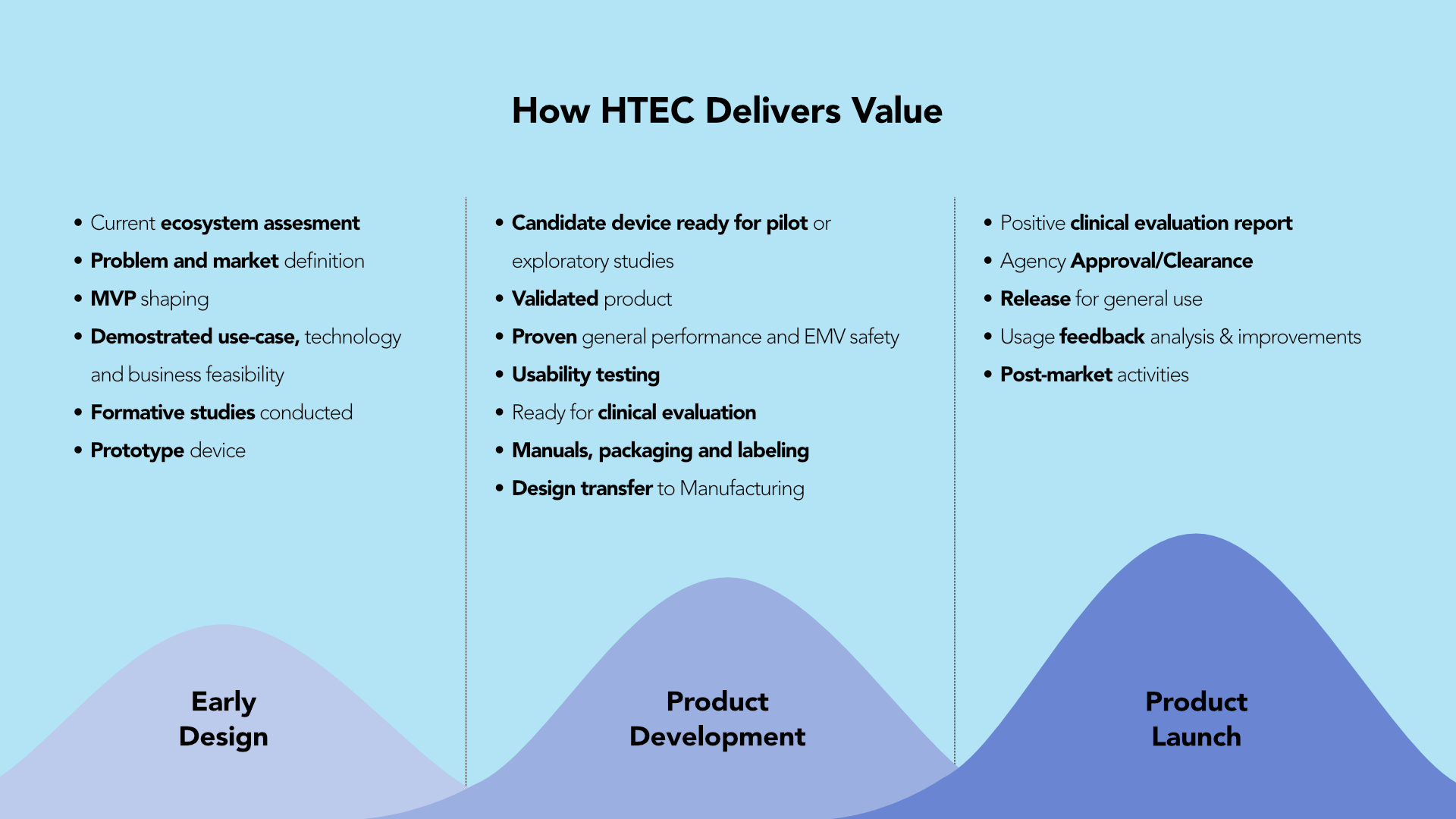
“A small piece of advice given early on can help clients avoid overinvesting in efforts that are not required at all.”
Optimism for the future: propelling next-generation medical devices
There are many other exciting developments happening in the field of MedTech and Biodesign that have the potential to improve healthcare and benefit society.
“We cannot go further without mentioning wearables and smart devices that can provide real-time feedback to help people make healthier lifestyle choices and offer a holistic view to care professionals about an individual before the illness. In biomedical engineering for example, researchers are working on developing new materials and methods for creating artificial organs, which could help address shortages of organs for transplantation. A lot is going on in telemedicine after COVID. Telemedicine has the potential to increase access to healthcare for people who live in remote or underserved areas, and to reduce the burden on healthcare systems by enabling patients to receive care from their own homes.”
Finally, Milos explains that there is “a huge potential in personalized medicine. With advances in genomics and data analysis, we can enable healthcare providers to tailor treatments and prevention strategies to the specific needs and characteristics of individual patients. This could lead to more effective and efficient healthcare, with better outcomes for patients. Overall, these and other developments in MedTech and biodesign have the potential to significantly improve healthcare and make it more accessible and affordable for people around the world.”
Milos also adds that countries from this region, Serbia, Bosnia and Herzegovina and Montenegro are not subjected to GDPR and clinical trials in this part of Europe are substantially cheaper and far less complex to conduct.
“We have a chance to leverage this opportunity to build ground-breaking medical solutions supported by some of the best experts and medical institutions in the region.”
>>>
To learn more about how we can help you build transformative medical solutions and contribute to better care for all, talk to our experts or connect with Milos Cigoj. In the meantime, check out our success stories to see how we help provide better health for all.